AlphaFold2: Wie ein Algorithmus fünfzig Jahre Biologie überholte
November 2020. Ein Algorithmus tritt gegen die besten Strukturbiologen der Welt an. Das Ergebnis erschüttert eine ganze Wissenschaft – und bringt 2024 den Nobelpreis.
Stell dir vor, du hast einen Bauplan. Buchstaben auf einem Blatt Papier. Zwanzig verschiedene Zeichen, aneinandergereiht wie Perlen auf einer Schnur. Tausende davon. Und aus diesem Bauplan soll eine Maschine entstehen. Eine Maschine so komplex, dass sie Krankheiten heilen, Gifte neutralisieren oder Signale durch deinen Körper senden kann.
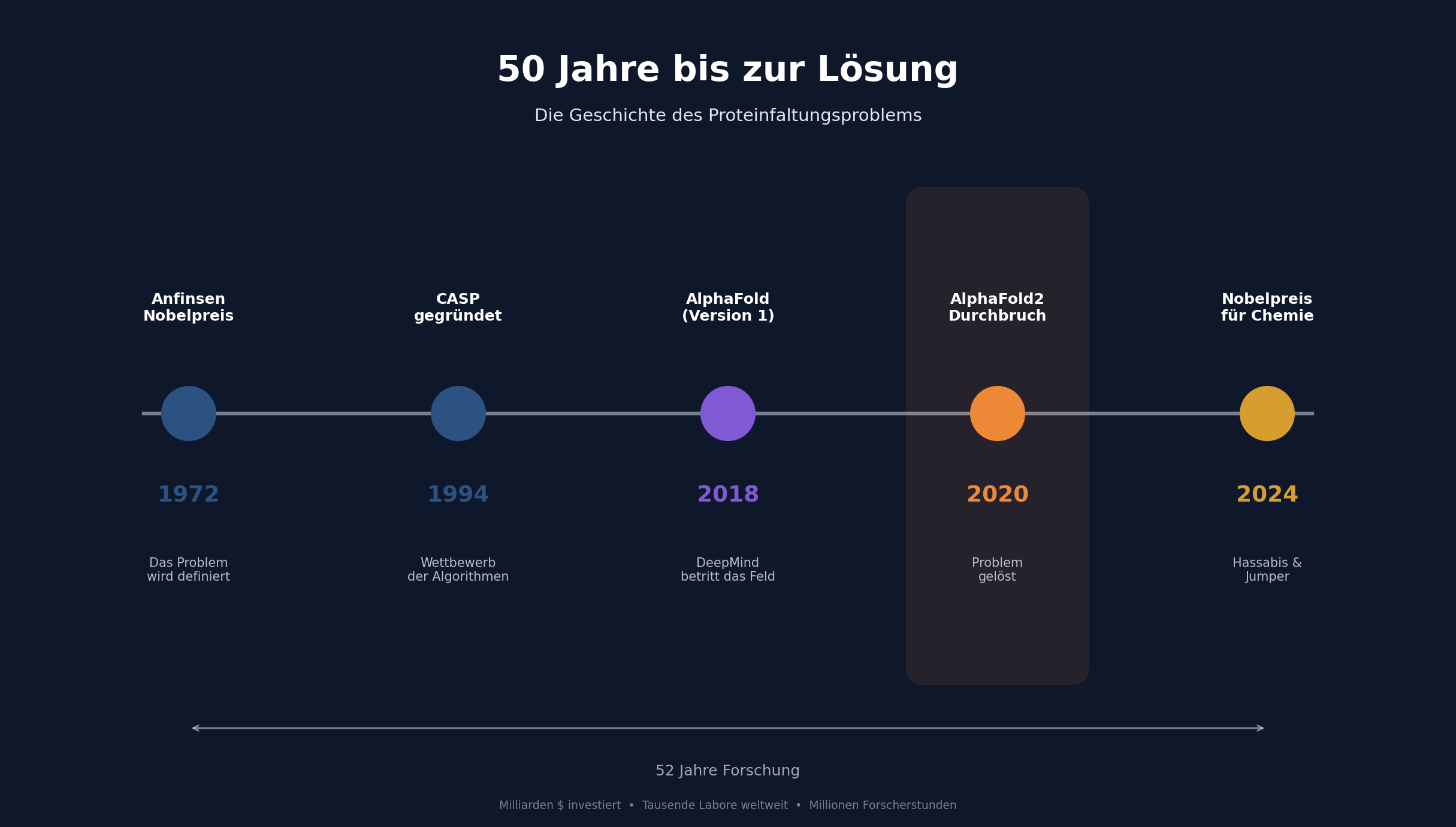
Das ist ein Protein. Und die Frage, wie aus der Buchstabenkette die dreidimensionale Maschine wird, beschäftigte Wissenschaftler seit 1972. Fünfzig Jahre. Tausende Labore weltweit. Milliarden an Forschungsgeldern.
Dann kam der November 2020.
Das Problem, das niemand lösen konnte
Christian Anfinsen bekam 1972 den Nobelpreis für eine Entdeckung: Die Reihenfolge der Aminosäuren bestimmt die Form des Proteins. Klingt einfach. Ist es nicht.
Ein durchschnittliches Protein besteht aus 300 Aminosäuren. Jede einzelne kann sich in verschiedene Richtungen drehen. Die Anzahl der möglichen Formen? Mehr als Atome im beobachtbaren Universum. Wenn ein Computer jede Möglichkeit eine Nanosekunde lang prüfen würde, bräuchte er länger als das Alter des Universums.
Und doch faltet sich jedes Protein in deinem Körper in Millisekunden. Perfekt. Jedes Mal.
Die Natur kennt den Trick. Die Wissenschaft nicht.
Experimentelle Methoden wie Röntgenkristallographie oder Kryo-Elektronenmikroskopie konnten einzelne Strukturen aufklären. Aber jede Struktur kostete Monate, manchmal Jahre. Und Millionen. Bis 2020 waren gerade einmal 48 Prozent des menschlichen Proteoms strukturell bekannt.
Der Wettbewerb der besten Köpfe
Seit 1994 gibt es CASP – Critical Assessment of protein Structure Prediction. Alle zwei Jahre treten Forschergruppen aus aller Welt gegeneinander an. Die Aufgabe: Proteinstrukturen vorhersagen, bevor die experimentellen Ergebnisse veröffentlicht werden. Ein Blindtest für Algorithmen.
Jahrelang bewegten sich die Ergebnisse im Bereich von 40 bis 60 Punkten auf der GDT-Skala. 100 wäre perfekt. 90 gilt als experimentell genau. Niemand kam auch nur in die Nähe.
Dann meldete sich ein neuer Teilnehmer an: DeepMind. Die Firma, die eine KI gebaut hatte, die den weltbesten Go-Spieler schlug. Diesmal wollten sie etwas anderes schlagen: ein halbes Jahrhundert Biologie.
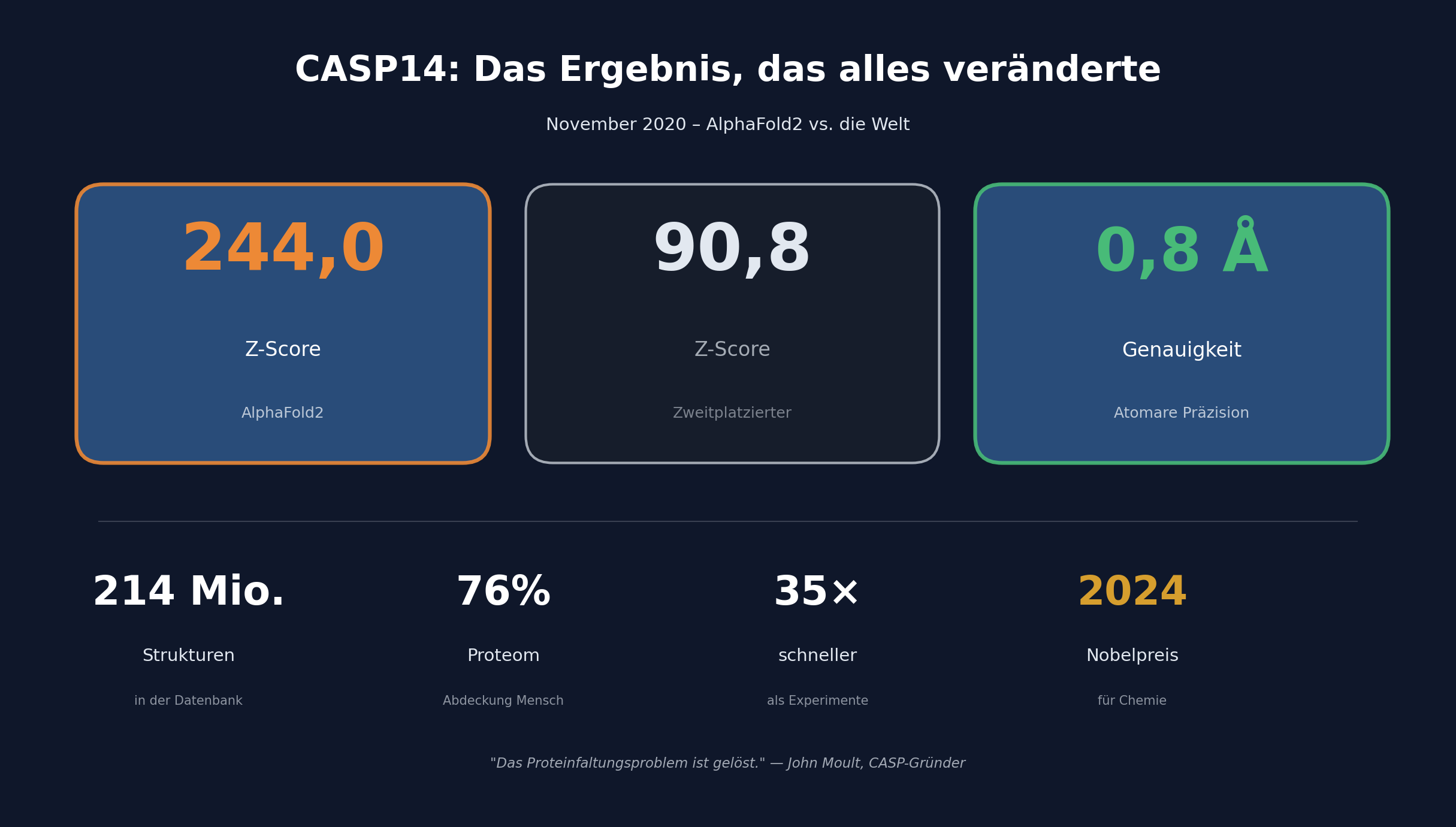
30. November 2020
Die Ergebnisse wurden bekannt gegeben. Stille im Raum. Dann Unglaube.
AlphaFold2 erreichte einen GDT-Score von 92,4. Bei manchen Proteinen lag die Abweichung bei 0,8 Ångström. Ein Ångström ist ein Zehnmilliardstel Meter. Die Breite eines Atoms.
Der zweitplatzierte Teilnehmer? 2,8 Ångström Abweichung. AlphaFold2 war nicht besser. Es war in einer anderen Liga.
Der summierte Z-Score, ein statistisches Maß für die Gesamtleistung: 244,0 für AlphaFold2. Das nächstbeste Team: 90,8. Fast dreimal so gut wie alle anderen. Und das nicht nur in einer Kategorie. AlphaFold2 gewann in Template-Based Modeling. Es gewann in Free Modeling. Es gewann überall.
„Das ist ein Erdbeben. Das Proteinfaltungsproblem ist gelöst."
Wie ein Algorithmus lernte, die Sprache des Lebens zu lesen
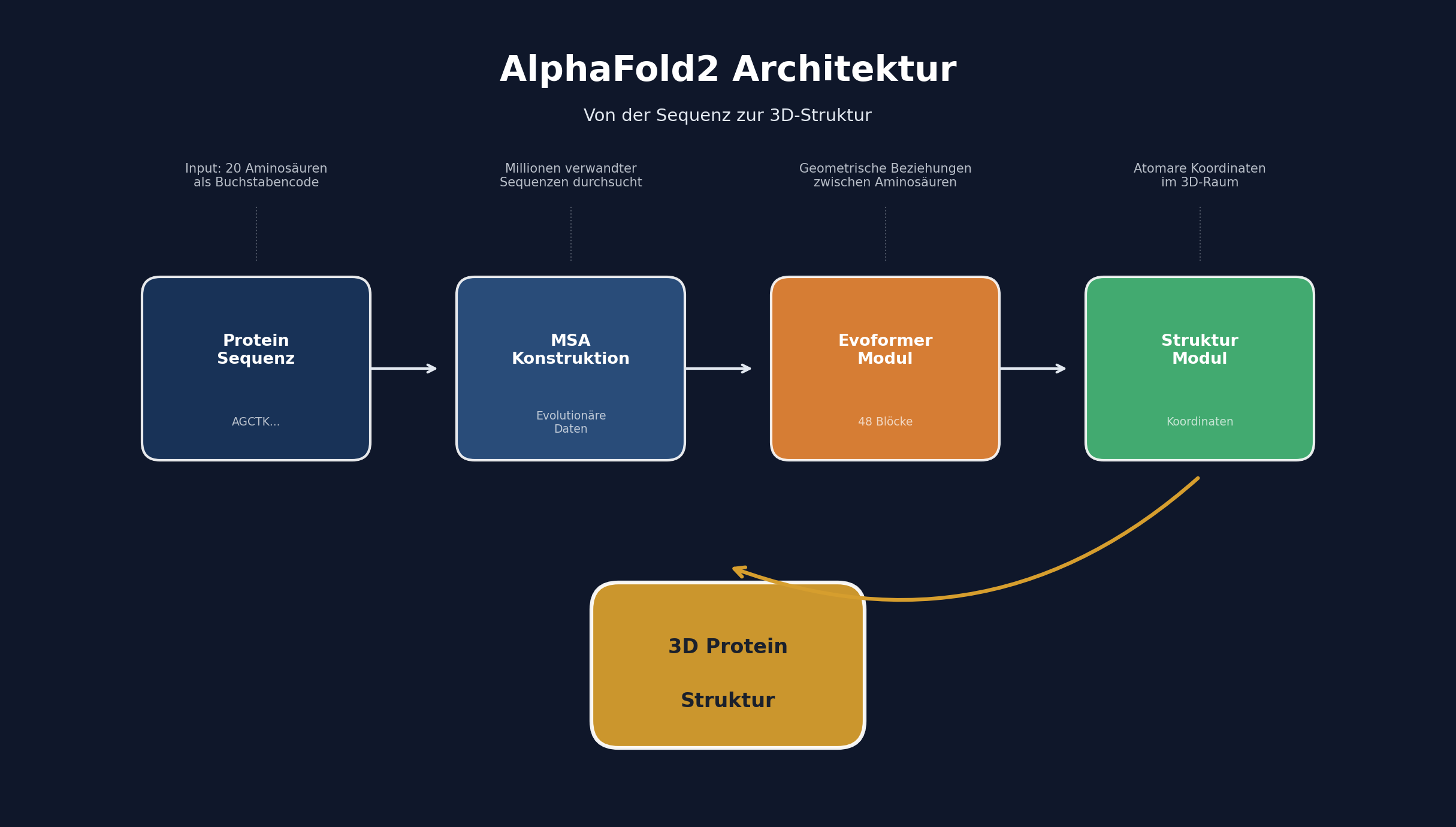
AlphaFold2 schaut nicht einfach auf eine Proteinsequenz. Es durchforstet Datenbanken mit Millionen von Sequenzen aus allen Lebewesen der Erde. Bakterien, Pflanzen, Tiere. Und es findet Muster.
Wenn zwei Aminosäuren über Millionen Jahre Evolution immer zusammen variieren, bedeutet das: Sie sind in der fertigen Struktur nahe beieinander. Die Evolution hinterlässt Spuren. AlphaFold2 hat gelernt, diese Spuren zu lesen.
Das Herzstück des Systems besteht aus zwei Modulen. Der Evoformer analysiert nicht nur Paare von Aminosäuren. Er betrachtet Dreiecke. Wenn A nahe bei B ist und B nahe bei C, was bedeutet das für A und C? Diese geometrische Logik zieht sich durch das gesamte Protein. Informationen fließen bidirektional zwischen der Mehrfach-Sequenzausrichtung und der Paar-Repräsentation – ein ständiger Austausch, der das Netzwerk immer präziser macht.
Das Struktur-Modul übersetzt dann diese abstrakten Beziehungen in echte Koordinaten im Raum. Atom für Atom. Bis die vollständige dreidimensionale Struktur steht.
214 Millionen Strukturen. Kostenlos.
DeepMind veröffentlichte nicht nur den Algorithmus. Sie berechneten die Strukturen für praktisch jedes bekannte Protein auf der Erde. 214 Millionen Strukturen. Alle frei zugänglich in der AlphaFold Protein Structure Database.
Die Zahlen sprechen für sich:
Vor AlphaFold2 waren 48 Prozent des menschlichen Proteoms strukturell bekannt. Danach: 76 Prozent. Von 5.027 menschlichen Proteinen ohne jegliche Strukturinformation blieben nur 29 übrig. Was experimentelle Strukturbiologie in fünfzig Jahren erreichte, übertraf AlphaFold2 in zwei Jahren.
Aber es blieb nicht dabei. Der ESM Metagenomic Atlas von Meta ergänzt diese Ressource mit 617 Millionen vorhergesagten Strukturen aus metagenomischen Daten – Proteine von Organismen, die noch nie ein Mensch kultiviert hat. Entdeckt in Bodenproben, Meeressedimenten, heißen Quellen.
Was das bedeutet
Ein Forscher in Cambridge nutzt AlphaFold2, um Proteine in Bienenvölkern zu verstehen. Er will herausfinden, warum manche Bienen gegen bestimmte Krankheiten resistent sind. John Jumper, einer der Entwickler von AlphaFold2, hatte das nie erwartet: „Ich hätte nie gesagt, dass AlphaFold für Bienenforschung verwendet wird."
Ein Labor in Basel beschleunigt damit die Entwicklung neuer Krebsmedikamente. Ein Team in Tokio analysiert Proteine von Viren, die noch nie jemand untersucht hat. Ein Student in Nairobi greift auf dieselbe Datenbank zu wie Nobelpreisträger in Harvard.
Während der COVID-19-Pandemie berechnete AlphaFold2 Strukturen neuer Virusvarianten in Stunden. Früher hätte das Monate gedauert. Die schnelle Verfügbarkeit der Spike-Protein-Struktur von SARS-CoV-2 war essentiell für die Entwicklung der mRNA-Impfstoffe. Die Anpassung an neue Varianten wurde dadurch erheblich beschleunigt.
Auch die experimentelle Strukturbiologie profitiert. AlphaFold2-Vorhersagen dienen als Startmodelle für Röntgenkristallographie und Kryo-Elektronenmikroskopie. Was früher der schwierigste Schritt war – ein initiales Modell zu finden – ist heute oft nur noch ein Klick.
Die Grenzen
AlphaFold2 ist kein Alleskönner. Es zeigt einen Schnappschuss. Proteine bewegen sich, verändern ihre Form, tanzen. Diese Dynamik erfasst das System nicht. Es ist ein Single-State-Vorhersager, ausgelegt auf eine einzelne, statische Struktur mit der niedrigsten freien Energie.
Manche Proteinbereiche haben keine feste Struktur. Sie sind von Natur aus ungeordnet, wie ein Faden im Wind. AlphaFold2 zeigt sie trotzdem gefaltet – eine kondensierte, oft falsche Faltung statt der inhärenten Unordnung. Ein bekannter Fehler.
Auch Punktmutationen, einzelne ausgetauschte Aminosäuren, sind problematisch. Die berechneten Unterschiede zwischen Original und Mutante sind oft minimal, typischerweise mit einem RMSD von weniger als 1 Ångström. In der Realität können die Auswirkungen gravierend sein. Für die Untersuchung genetischer Erkrankungen ist das eine echte Einschränkung.
Protein-Komplexe aus mehreren Untereinheiten bereiten ebenfalls Schwierigkeiten. AlphaFold-Multimer wurde entwickelt, um diese Limitation anzugehen, kämpft aber besonders bei Heteromeren – Komplexen aus unterschiedlichen Untereinheiten. Antigen-Antikörper-Interaktionen mit ihrer extremen Sequenzvariabilität bleiben eine besondere Herausforderung.
Die Konkurrenz holt auf
AlphaFold2 hat Nachfolger bekommen. ESMFold von Meta braucht keine zeitaufwendige Mehrfach-Sequenzausrichtung. Es ist 35-mal schneller. Die Genauigkeit ist etwas geringer – ein TM-Score von 0,68 statt 0,85 – aber für viele Anwendungen ausreichend. Besonders wenn keine evolutionären Verwandten bekannt sind, spielt ESMFold seine Stärken aus.
EMBER3D geht einen anderen Weg und zeigt seine Stärke bei der Analyse von Punktmutationen – genau dort, wo AlphaFold2 schwächelt.
Und dann ist da AlphaFold3. Die neueste Version erweitert die Fähigkeiten auf Protein-Komplexe, DNA, RNA und kleine Moleküle. Die Interaktion zwischen verschiedenen Biomolekülen, bisher ein blinder Fleck, rückt in den Fokus.
Der Nobelpreis
2024 erhielten Demis Hassabis und John Jumper den Nobelpreis für Chemie. Für ein Problem, das fünfzig Jahre niemand lösen konnte. Gelöst in vier Jahren intensiver Arbeit.
Es ist einer jener seltenen Momente, in denen die Wissenschaft anerkennt: Hier hat sich etwas fundamental verändert. Nicht inkrementell. Nicht graduell. Sondern radikal.
Was bleibt
Die AlphaFold Protein Structure Database ist unter alphafold.ebi.ac.uk erreichbar. Kostenlos. Für jeden. Ob du in einem Labor arbeitest oder einfach neugierig bist.
Forscher weltweit arbeiten an den Limitationen. Die AlphaFill-Datenbank transplantiert automatisch Liganden und Kofaktoren aus experimentellen Strukturen in AlphaFold2-Modelle. Neue Ansätze mit Netzwerkgewichte-Perturbation erkunden den konformationellen Raum, um alternative Strukturzustände zu finden.
Fünfzig Jahre Forschung. Milliarden investierte Dollar. Tausende Karrieren. Und am Ende kam die Lösung von einem Algorithmus, der die Sprache der Evolution lesen lernte.
Das Proteinfaltungsproblem ist gelöst. Die Arbeit beginnt gerade erst.
Quellen
- AlphaFold Protein Structure Database: alphafold.ebi.ac.uk
- Jumper, J. et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature.
- CASP14 Ergebnisse: predictioncenter.org
- Frontiers in AI: "An Overview of AlphaFold's Breakthrough"
- Nobelpreis für Chemie 2024: nobelprize.org